Dibenzo[b,j][1,10]
phenanthrolines are an interesting group of compounds. They have the potential
to combine the properties of Polycyclic Aromatic Hydrocarbons
(PAHs) with the flexibility of coordination complexes. The compounds have been
used in macrocycles to inhibit telomerases and destabilize DNA, can activate
nucleases when used in copper (II) complexes, and have been shown to readily
form coordination complexes with ruthenium [1-4]. The ruthenium complex could be used as a sensitizing dye to improve the
efficiency of the Dye-Sensitive
Solar Cells (DSSCs) and others. However, the current applications of
dibenzo[b,j][1,10]phenanthrolines are rather limited, potentially because of
the low synthetic yield. Introduction of two methyl groups on dibenzo[b,j][1,10]phenanthrolines, i.e.
synthesis of 5,8-dimethyl-dibenzo[b,j][1,10]phenanthroline (compound 1, Scheme 1), is an important step in the
applications of this type of chemicals since the methyl can be readily
converted to other functional groups, such as -CH2Cl, -COOH, for the synthesis
of dibenzo[b,j][1,10] phenanthrolines derivatives.
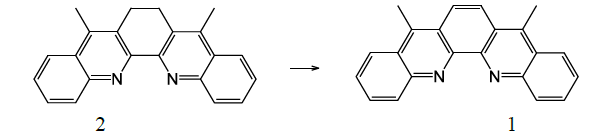
Scheme 1:
Palladium-catalyzed dehydrogenation to produce
5,8-dimethyl-dibenzo[b,j][1,10]phenanthroline (1).
Several
methods have been reported on the synthesis of 1 and its relatives, and the
best method appeared to be the one reported by Kempter and Stoss according to
the yield. It involved a Friedländer
condensation, followed by a palladium-catalyzed dehydrogenation of the
product [5-12].
The
Friedländer condensation proceeded with high yields and no difficulty, but
yields from literature for the palladium-catalyzed dehydrogenation from 2
(Scheme 1) were quite low (10-20%). Conditions for this reaction relied on
elevated temperatures (210°C), which have a high potential for unwanted side
reactions on a compound of this type, potentially explaining the low yield. No
conversion was seen at lower temperatures in methanol, toulene, xylenes,
1,2-dichlorobenezene, acetic acid, and decalin. Decomposition was seen
in nitrobenzene, and limited yield (10%) was seen in isocetane. This indicates
a high activation energy barrier for the reaction.
Dehydrogenation
to introduce an olefinic bond can be done via oxidation with a number of
different standard oxidizing
agents. However, all of our attempts on similar systems to generate
compound 1 from 2 have failed. Another more effective method of dehydrogenation
via bromination/dehydrobromination
under mild conditions was pioneered by Barnes in 1948.However, since the
benzylic positions at C6/C7 and the methyl groups are
equally reactive, making 1 directly from 2 via this process was not successful
unless the methyl is protected, and subsequent reduction is also effective on
the central bond [13-16].
In this work, we report a novel two-step reaction with a total 63% yield
starting from 1,2-dibromobenzene and 2aminoacetophenone (Scheme 2). The first step is a Buchwald-Hartwig amination.
Our results show that when 2,2-bis(diphenylphosphino)-1,1binaphthyl (BINAP) was
used,reaction yields up to 60% of 3 were achieved, with
2-dicyclohexylphosphino-2,4,6-triisopropylbiphenyl (XPhos) proving far more
successful, with yields>80% under the proper conditions. The reaction was
optimized using a procedure developed for this ligand in Buchwalds lab, using
stoichiometric amounts of water to activate the Pd(OAc)2/XPhos
precatalyst system [17-19].
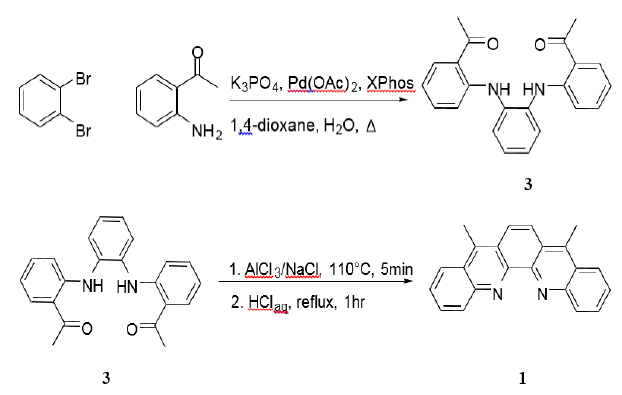
Scheme 2:
Buchwald-Hartwig amination of 1,2-dibromobenzene with 2-aminoacetophenone to 3
and 6-exo-trig ring closing reaction of 3 to 1.
This
allowed a 90% yield and the reaction time to be accelerated from 4-6 days using
the Ullmann reaction
to overnight [20]. NMR analysis of the reaction mixture shows that the
individual displacements behave semi-sequentially, with monosubstitution being
preferentially followed by monosubstitution of another molecule, rather than
disubstitution of the same. It is noteworthy that attempts to use (tBu)3P
as the ligand failed, as β-arylation proved to be an inescapable byproduct from
the reactive acetyl functionalities.
The
second step is the ring-closing reaction of 3. A synthesis listed in a paper
published in 2012 seemed promising, as it used a targeted Lewis acid (AlCl3)
along with a strong Brönsted acid. In the past, every substrate to date using
this combination was a substituted anthraquinone of some kind, which have very
different properties from diphenyl ketones or diphenylamines [21-24]. However,
this reaction still turned out to be quite successful for the synthesis of 1.
The reaction starts with a eutectic mixture of AlCl3 and NaCl that
melts at 110°C. This dissolves
the substrate, and can then be carefully diluted with 4M HCl to finish the
reaction (Scheme 2). Yields were surprisingly good (70%), and the reaction
yielded a clean product without chromatography.
Although many acids have been used to crosslink acetophenone with benzene
rings, most of them proved unsuccessful in making 1. Phosphoric acid, sulfuric
acid, and Eatons reagent produced decomposition and a complicated mixture of
products. Acetic acid catalyzed by sulfuric acid caused the formation of
carboxylic acid byproducts before the desired target could be formed [25-29]. Trifluoroacetic acid
formed an unidentified product upon reaction with 3. Weak Lewis acids such as
In(III), Sn(II), and even Fe(III) also produced no results. Imidazolium-based
ionic liquids and deep eutectic salts, such as that formed between choline
chloride and ZnCl2, failed to react as well.
Complexation
reactions to make a Ruthenium complex are chemically quite straightforward. A
metal precursor is chosen with coordinating ligands sufficiently labile to be
displaced by the incoming nucleophile. Minimum necessary conditions were tested
by mixing one equivalent of 1 with one equivalent of Ru(DMSO)4Cl2
in polar solvent systems with ever increasing boiling points. While 2,2-bipyridine-4,4-dicarboxylic
acid (dcbpy) would readily coordinate in as mild conditions as refluxing
1,2-dichloroethane, 1 failed to coordinate until refluxing
N,N-dimethylformamide (DMF). Even then, coordination was so slow as to take
multiple days, so conditions were increased to ethylene glycol. In hot (170°C)
ethylene glycol, the coordination takes less than 30 minutes. Therefore, these
conditions were used to create the chloride salt of the final ruthenium
complex, with a final reaction in DMF to make the thiocyanate (Scheme 3).
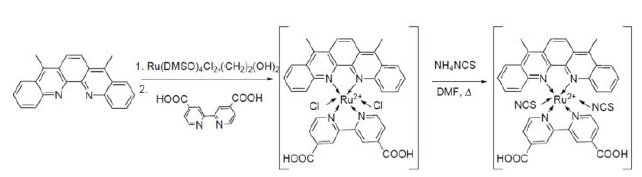
Scheme 3: Coordination of
1 to its ruthenium complex.
The
DSSC devices based on this Ru complex will be studied and reported in the
future. Also, further synthesis could create a library of derivatives that
could yield much more insight into this system. This could be used to compare
the effect of planarity on the efficiency of the solar cell. By comparing
dibenzophenanthroline, dihydrodibenzophenanthroline, and biquinoline
derivatives, a more complete picture could be drawn as to the interaction of
highly aromatic compounds in DSSCs.
Experimental
All
reactions were performed under nitrogen unless specified otherwise. All
chemicals were purchased from Fisher Scientific and were used as received.
Deuterated solvents were purchased from Acros Organics. Nuclear Magnetic Resonance
(NMR) spectra were obtained on Varian INOVA 300 MHz and 500 MHz NMR
spectrometers. Mass
spectrometry (HRMS) experiments were conducted using Micromass AutoSpec M
magnetic sector using Chemical Ionization (CI) in methane.
1,1-((1,2-phenylenebis(azanediyl))bis(2,1-phenylene))
bis(ethan-1-one) (3)
Into
a round bottom was added palladium (II) acetate (6.6mg, 29.4 μmol), Xphos (41.7
mg, 87.4 μmol), and potassium phosphate (921 mg, 4.34 mmol), and an inert
atmosphere was established. 3 mL of dioxane with 2 μL of water were then added
to the round bottom, and the orange solution was heated until the color had
deepened to a dark red. The 1,2-dibromobenzene (327.4 mg, 1.39 mmol) and 2-aminoacetophenone
(420.6 mg, 3.11 mmol) were then added along with an addition 3 mL of dioxane,
and the reaction was heated to reflux overnight. The dioxane was removed in the
rotary evaporator, and the reaction mixture was resuspended in dichloromethane
with added Celite. This was filtered and washed with an additional 20 mL of
dichloromethane.
The
dark red solution was then washed with 3 portions of 30 mL 0.8M HCl to remove
the excess amine, followed by 2 portions of 30 mL of water. The resulting
brownish solution was dried over magnesium sulfate, rotovapped, and
chromatographed on silica (30:70 ethyl acetate:hexanes) to yield 428.8 mg of
yellow solid (90%). 1H NMR (499 MHz, Chloroform-d) δ10.33 (s, 2H), 7.73 (dd, J=8.1,
1.6 Hz, 2H), 7.43 (dd, J=5.9, 3.6 Hz, 2H), 7.27-7.21 (m, 2H), 7.14 (dd, J=5.9,
3.6 Hz, 2H), 7.03 (dd, J=8.5, 1.1 Hz, 2H), 6.69 (ddd, J=8.1, 7.0, 1.1 Hz, 2H),
2.55 (s, 6H). MS 344.1.
5,8-dimethyldibenzo[b,j][1,10]phenanthroline
(1)
A
round-bottomed flask was placed in a nitrogen glove box and loaded with 21.245
g of a finely ground 4:1 mixture (by weight) of dried AlCl3 and
NaCl, respectively. This was then heated at 110°C until a clear
liquid was formed, and then cooled. 2.043 g (5.94 mmol) of 3 was added, and the
mixture was re-heated until a dark brown solution was formed. This was heated
for 5 minutes before cooling. Finally, 150mL of 4M HCL was slowly added until
the solution was completely neutralized, and it was reheated for 1 hr. The
resulting solution was filtered, poured on ice, slowly neutralized with NaHCO3,
and filtered again to collect the precipitate. The filter cake was washed with
water and cold methanol to yield 1.283 mg (4.17 mmol) of yellow solid (70.2%).
This
was then recrystallized from CH2Cl2/MeOH to yield yellow
needles. 1H NMR (498 MHz, Chloroform-d) δ8.70 (d, J=8.5 Hz, 2H), 8.31 (d, J=8.7
Hz, 3H), 8.09 (s, 2H), 7.87 (t, J=7.5 Hz, 2H), 7.70 (t, J=7.6 Hz, 2H), 3.16 (s,
6H). MS 308.1.
Ru(1)(dcbpy)NCS2
Ru(DMSO)4Cl2
(241.3 mg, 0.499 mmol) was added to a round-bottomed flask, along with 155.2 mg
(0.504 mmol) of 1 and 20 mL of dry ethylene glycol. This was heated at 170°C
for 30 min. 4,4-dicarboxyl-2,2-bipyridine (125.3 mg, 0.514 mmol) was then
added, and heating continued for another 2 hours. Finally, 253.2 mg (3.33 mmol)
of NH4NCS was added, and heating continued for another 3 hours. Once
the reaction was cooled, it was diluted with 0.1 M HNO3, and placed
at 4°C overnight. The resulting deep
red precipitate was filtered, washed with water and acetone, and dissolved in
0.1M Na2CO3. This was then re-precipitated with HNO3,
filtered, and washed again with water and acetone to yield 174.1 mg of red
solid (45%) 1H-NMR (500 MHz, DMSO-d6) d 10.04 (d, J=7.9 Hz, 1H), 9.91 (d, J=8.8
Hz, 1H), 8.83 (d, J=3.1 Hz, 1H), 8.82 (d, J=1.6 Hz, 1H), 8.57 (d, J=3.2 Hz,
1H), 8.56 (d, J=0.6 Hz, 1H), 8.46 (d, J=5.4 Hz, 1H), 8.21 - 8.18 (m, 2H), 7.73
(d, J=5.4 Hz, 1H), 7.47 (s, 1H), 7.28-7.21 (m, 2H), 7.20-7.11 (m, 2H), 6.18 (d,
J=8.2 Hz, 1H), 3.90 (s, 6H).
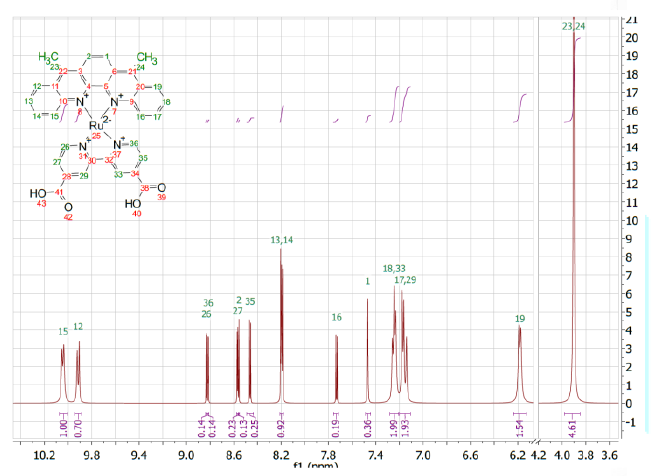
f1 ppm
References
1.
Artese
A, Costa G, Distinto S, Moraca F, Ortuso F, et al. Toward the design of new DNA
G-quadruplex ligands through rational analysis of polymorphism and binding data
(2013) Eur J Med Chem 68: 139-149. https://doi.org/10.1016/j.ejmech.2013.07.022
2.
Teulade-Fichou
MP, Fauquet M, Baudoin O, Vigneron JP and Lehn JM. DNA double helix
destabilizing properties of cyclobisintercaland compounds and competition with
a single strand binding protein (2000) Bio org Med Chem 8: 215-222. https://doi.org/10.1016/s0968-0896(99)00283-7
3.
Baudoin
O, Fichou MPT, Vigneron JP and Lehn JM. Efficient copper (II)-mediated nuclease
activity of ortho-quinacridines (1998) Chem. Commun. 2349-2350. https://doi.org/10.1039/a806095i
4.
Belser
P and Zelewsky A. Synthese, spektroskopische eigenschaften und
elektrochemisches verhalten von ruthenium(ii)‐komplexen mit zweizähnigen
stickstoffliganden (1980) Helvetica Chimica Acta 63: 1675-1702. https://doi.org/10.1002/hlca.19800630637
5.
Heitwinkel
D and Ittemann P. Eine allgemeine Synthesemethodefür Dibenzo[b,j] [x,z]phenanthroline
mit x,z=1,7; 4,7 und 1,10 (1985) Liebigs Ann Chem 1501 - 1507.
https://doi.org/10.1002/jlac.198519850722
6.
Lartia
R, Bertrand H and Teulade-Fichou MP. Improved synthesis of quinacridine
derivatives (2006) Synlett 4: 610-614. https://doi.org/10.1055/s-2006-932465
7.
Wu
FY and Thummel RP. Recent Advances in the Friedländer Reaction Chemical Reviews
(2002) Inorg Chim Acta 327: 26-30.
8.
Jahng
Y, Hazelrigg J, Kimball D, Riesgo E, Wu F, et al. Copper(I) complexes of 3,3-bridged
2,2-biquinoline: synthesis, properties, and structure (1997) Inorg Chem 36:
5390-5395. https://doi.org/10.1021/ic970385v
9. Madureira
J, Santos TM, Goodfellow BJ, Lucena M, Pedrosa de Jesus J, et al. Structural
characterisation of new RuII[9]aneS3 polypyridylic complexes (2000) J Chem Soc
Dalton Trans 4422-4431. https://doi.org/10.1021/ic970385v
10. Klimant
I, Belser P and Wolfbeis OS. Novel metal-organic ruthenium(II) diimin complexes
for use as longwave excitable luminescent oxygen probes (1994) Talanta 41:
985-991. https://doi.org/10.1016/0039-9140(94)e0051-r
11.
Kempter
G and Stoss W. Heterocyclen aus aminoketonen (1963) Z Chem 3: 61-62. https://doi.org/10.1002/zfch.19630030206
12.
Kempter
G and Stoss W. Heterocyclen aus Aminoketonen. II. Reaktionen mit alicyclischen
1,2-Dionen (1963) J Prakt Chem 21: 198-203. https://doi.org/10.1002/prac.19630210312
13.
YoshidaM,
NagayamaS, MinabeM and SuzukiK. Electrophilic substitution of
4H-cyclopenta[def]phenanthrene. Nitration (1979) J Org Chem 44: 1915-1917. https://doi.org/10.1021/jo01326a004
14.
Minabe
M, Yoshida M and Kimura O. Synthesis of 2,6,8-trinitro-4h-cyclopenta[def]
phenanthren-4-one as an electron acceptor (1985) Bull Chem Soc Jpn 58: 385-386.
https://doi.org/10.1002/chin.198529174
15. Míšek
J, Teplý F, Stará I, Tichý M, Šaman D, et al. A straightforward route to
helically chiral n-heteroaromatic compounds: practical synthesis of racemic
1,14-diaza[5]helicene and optically pure 1- and 2-aza[6]helicenes (2008) AngewChem
Int Ed 47: 3188-3191. https://doi.org/10.1002/anie.200705463
16.
Barnes
RA. N-bromosuccinimide as a dehydrogenating agent (1948) J Am Chem Soc 70: 145-147.
17.
Guram
AS, Rennels RA and Buchwald SL. A simple catalytic method for the conversion of
aryl bromides to arylamines (1995) Angew Chem Int Ed 34: 1348-1350. https://doi.org/10.1002/anie.199513481
18. Louie
J and Hartwig JF. Palladium-catalyzed synthesis of arylamines from aryl
halides. mechanistic studies lead to coupling in the absence of tin reagents (1995)
Tetrahedron Lett 36: 3609-3612. https://doi.org/10.1016/0040-4039(95)00605-c
19.
Fors
BP, Krattiger P, Strieter E and Buchwald SL. Water-mediated catalyst
preactivation: an efficient protocol for c-n cross-coupling reactions (2008)
Org Lett 10: 3505-3508. https://doi.org/10.1021/ol801285g
20.
Ullmann
F and Sponagel P. Ueber die Phenylirung von Phenolen (1905) Ber Dtsch Chem Ges
38: 2211-2212. https://doi.org/10.1002/cber.190503802176
21.
Park
BS, Lee SW, Kim IT, Tae JS and Lee SH. Synthesis and photoluminescent
properties of new ceramidine derivatives (2012) Heteroat Chem 23: 66-73. https://doi.org/10.1002/hc.20753
22.
Cook
AH and Waddington W. Experiments in the coeroxene and coeramidine series (1945)
J Chem Soc 1945 402-405. https://doi.org/10.1039/jr9450000402
23.
Sokolyuk
NT and Pisulina LP. Synthesis and photoarylotropic rearrangement of
10-phenoxynaphthacenokeramidonine (2002) Russ J Org Chem 38: 1212-1213.
https://doi.org/10.1002/chin.200318121
24.
Waldmann
H and Hindenburg KG. Über ms‐benzacridan-abkömmlinge (1940) J Für Prakt Chem
156: 157-168.
https://doi.org/10.1002/prac.19401560405
25.
Sugasawa
T, Toyoda T, Adachi M and Sasakura K. Aminohaloborane in organic synthesis. 1.
Specific ortho substitution reaction of anilines (1978) J Am Chem Soc 100:
4842-4852. https://doi.org/10.1021/ja00483a034
26. Ruediger EH,
Kaldas ML, Gandhi SS, Fedryna C and Gibson MS. Reactions of
1,5-dichloroanthraquinone with nucleophiles (1980) J Org Chem 45: 1974-1978. https://doi.org/10.1021/jo01298a044
27.
Shidlovskii
AF, Golubev AS, Gusev DV, Suponitsky KY, Peregudov AS, et al. A new synthesis
of N-substituted о-trifluoroacetylanilines (2012) J Fluor Chem 143: 272-280. https://doi.org/10.1002/chin.201311066
28.
HanXD,
ZhaoYL, MengJ, RenCQ and LiuQ. Synthesis of acridines and persubstituted
phenols from cyclobutenones and active methylene ketones (2012) J Org Chem 77:
5173-5178. https://doi.org/10.1021/jo300615t
29.
Kuninobu
Y, Tatsuzaki T, Matsuki T and Takai K. Indium-catalyzed construction of
polycyclic aromatic hydrocarbon skeletons via dehydration (2001) J Org Chem 76:
7005-7009. https://doi.org/10.1021/jo200861s
*Corresponding author
Hai-Feng Ji, Department of Chemistry, Drexel University,
Philadelphia, 19104, United states, E-mail: hj56@drexel.edu
Citation
Johnson MN and Ji HF. Synthesis of
5,8-dimethyl-dibenzo[b,j][1,10]phenanthroline and its Ru complex (2019)
Nanomaterial Chem Technol 1: 36-39.



